
Premium Eyes Augenlasern unter der Leitung von Focus-Top-Mediziner Dr. Detlev R.H. Breyer ist Ihr Zentrum für Brillenunabhängigkeit durch sanftes Augenlasern bei Kurzsichtigkeit, Weitsichtigkeit, Hornhautverkrümmung und Alterssichtigkeit sowie Linsenverfahren bei grauem Star.

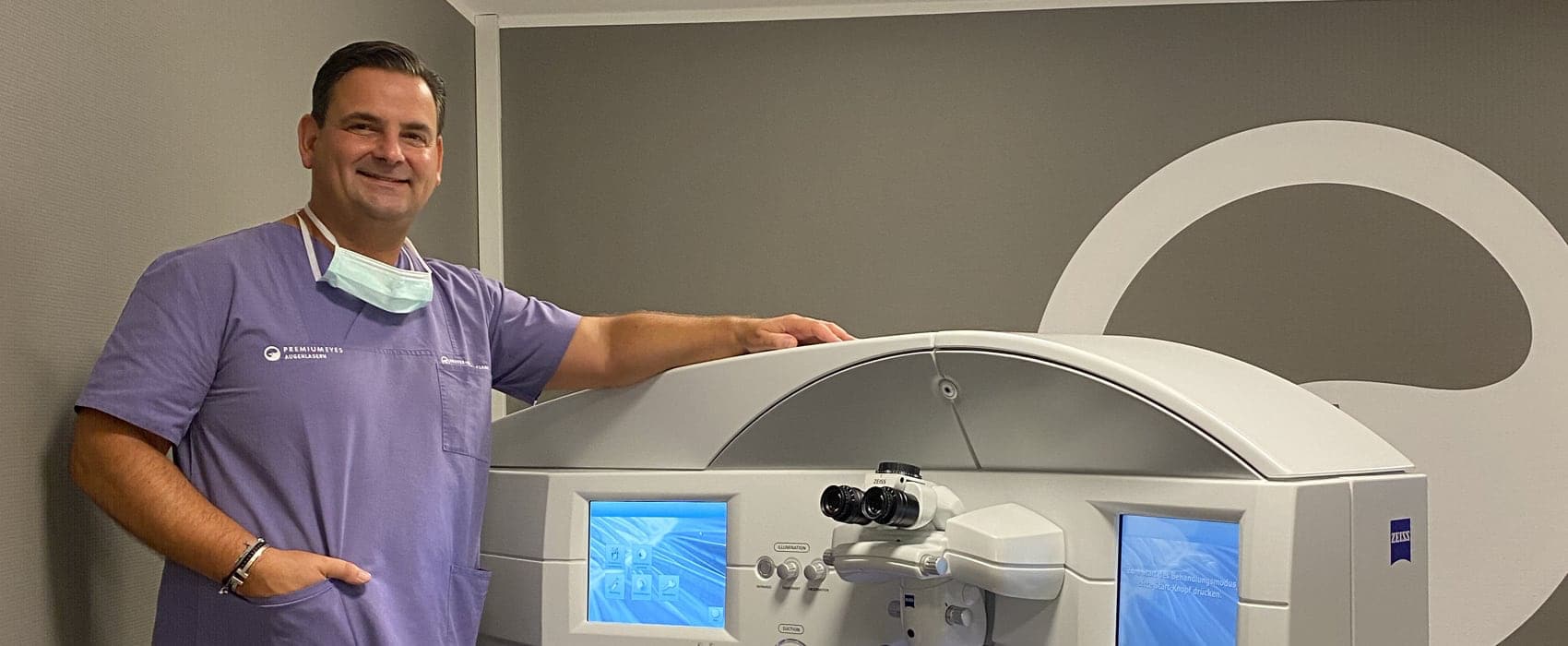
Individuelle Therapien sowie schonende und schmerzfreie Augenoperationen sind Leidenschaften unserer Augenärzte
Um Ihre Augen schonend und schmerzfrei zu behandeln oder zu operieren, führen unsere Augenärzte als Kooperationspartner im IVCRC.net der UniversitätsAugenklinik Heidelberg eigene Forschungsprojekte durch. In unserem Team von Augenärzten mit unterschiedlichen Spezialisierungen finden auch Sie einen erfahrenen Augenarzt oder Augenchirurgen in Düsseldorf. Wir sind gerne für Sie da. – Möchten Sie unabhängig von Brille, Lesebrille oder Kontaktlinsen werden? Dann sind Sie unserem Augenlaserzentrum Premium Eyes genau richtig. Behandlungen von Makula, Netzhaut und Glaskörper bieten wir Ihnen in unserem Makula-Netzhaut-Zentrum in Düsseldorf-Oberkassel.

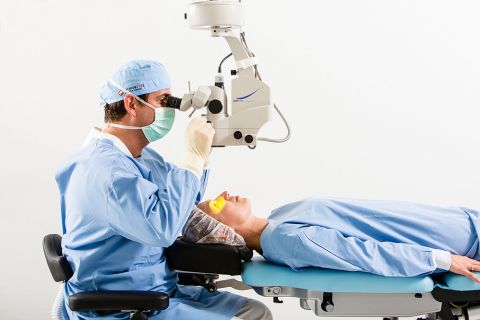

Hotlines für kurzfristige OP- und Behandlungstermine
Sie planen eine Grauer-Star-Operation, die Therapie von altersbedingter Makuladegeneration (AMD) im Makula-Netzhaut-Zentrum (MVZ) oder eine Augenlaser-OP bei Premium Eyes? Dann rufen Sie unsere Hotlines an oder buchen Sie online. Wir vergeben auch zeitnahe OP-Termine.

Neueste Innovationen
Moderne Laserbehandlungen für Ihre Augen

Schonende Augenlaserbehandlungen sind ein Spezialgebiet unserer Augenärzte, um Ihnen Unabhängigkeit von (Lese-)Brille oder Kontaktlinsen zu bieten und als Therapie bei vielen Augenerkrankungen. Hier finden Sie eine Übersicht über unsere innovativen Laserbehandlungen.





Unser Kompetenznetzwerk
Aufgrund ihrer überdurchschnittlichen fachlichen Qualifikation sind unsere leitenden Operateure ausgezeichnete und eingeladene Mitglieder nationaler und internationaler Fachgesellschaften. Sie sind Reviewer zahlreicher internationaler Fachzeitschriften. Darüber hinaus sind wir Mitglieder in den Netzwerken von Premium Praxen und von Primo Medico. Erfahren Sie mehr zu unserer Forschungstätigkeit.


















